Opis choroby
- Choroba Pompego (glikogenoza II)
- Klasyfikacja
- Częstotliwość występowania
- Typy choroby Pompego
- Rola lizosomu
- Genetyka
- Historia choroby Pompego
- Diagnozowanie choroby Pompego
- Testy laboratoryjne
Choroba Pompego jest rzadką, osłabiającą mięśnie
chorobą, dotykającą zarówno dzieci jak i dorosłych.
Wyróżniamy 2 typy choroby: występująca u dzieci (pojawiająca się
w ciągu kilku miesięcy po przyjściu na świat) oraz typ atakujący dzieci
starsze i dorosłych (przebieg stopniowy). Oba typy mogą być
charakteryzowane jako: postępujący zanik mięśni oraz trudność
oddychania. Przebieg choroby zależy od wieku, w którym pojawiła
się u chorego oraz od stopnia zaatakowania
organów. W typie choroby występującym u
dzieci obserwuje się powiększenie mięśnia sercowego. Ludzie,
którzy urodzili się z tą chorobą mają wrodzony niski
poziom enzymu zwanego kwasem alfa - glukoidalny GAA. Enzym ten
(białkowe molekuły w obrębie komórek) inicjuje biochemiczne
reakcje ludzkiego organizmu. U zdrowego człowieka (z prawidłowym
poziomem GGA) enzym ten uczestniczy w rozbijaniu glikogenu w obrębie
lizosomu. W przypadku choroby Pompego praca GGA może być osłabiona,
mieć nieprawidłowy przebieg lub w ogóle nie występować z powodu
nadmiaru nagromadzenia glikogenu w lizosomie.
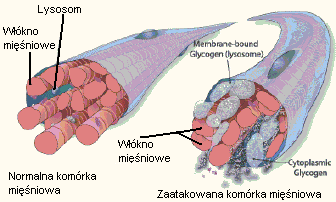
Rysunek po stronie prawej przedstawia sytuację, która występuje w przypadku, gdy glikogen jest akumulowany w zaatakowanych chorobą komórkach, co powoduje powiększenie lizosomu i prowadzi do ograniczenia funkcji mięśnia. W późniejszym stadium choroby lizosom może pęknąć uwalniając glikogen. Mięsień sercowy i kręgosłupa są zazwyczaj najbardziej zaatakowane.
Choroba Pompego jest określana jako:
- Metaboliczna choroba mięśni - dzieli objawy (takie jak zanik mięśni) z 40 innymi tego typu zaburzeniami (np. dysgrafia mięśni)
- Gromadzenie w obrębie lizosomu (Lysosomal storage disease) LSD - choroba zakłóca zdolność organizmu do rozbijania złożonych cząsteczek w obrębie lizosomu, powodując nadmierne nagromadzenie i jego nieprawidłowe funkcjonowanie
- Gromadzenie glikogenu (Glycogen storage disease) GSD - zmniejszenie zdolności organizmu do rozbijania i przetwarzania glikogenu.
Nie wiadomo dokładnie ile osób cierpi na tą chorobę. W przybliżeniu występuje ona u jednej osoby na 40000 urodzeń. Choroba Pompego jest chorobą rzadko występującą, dotykającą mniej niż 200000 ludzi w Stanach Zjednoczonych i nie więcej niż 5000 do 10000 w Europie. Choroba ta występuje u wszystkich ras ludzkich, z większą częstotliwością jednak u Afro-Amerykanów (1 na 14000 urodzeń), z mniejszą u Kaukazów (1 na 60000 dorosłych i 1 na 100000 dzieci).
W zależności od tego, kiedy pojawiają się pierwsze symptomy, chorobę
klasyfikujemy jako; dziecięcą, młodzieżową i wieku dorosłego. Symptomy
różnią się znacznie w zależności od kategorii wiekowej. Co
więcej, zauważono, iż zarówno wiek, w jakim pojawiła się choroba
u danej osoby, jak i stopień zaatakowania organów mają wiele
wspólnego ze sprawnością funkcjonowania enzymu GAA: u niemowląt
wynosi ona poniżej 1 % normalnej aktywności, u młodzieży poniżej 10%, u
dorosłych mniej niż 40%. Problemy z sercem są zależne od poziomu
aktywności enzymu GAA. Często występują u niemowląt, gdzie GAA jest
bardzo niski. U starszych chorych poziom GAA jest wyższy, więc problemy
z sercem występują znacznie rzadziej. Mięsień sercowy nie jest
atakowany.
Oszacowano, że 1/3 chorych cierpi na wczesną (pojawiającą się w
dzieciństwie) formę choroby. U osób, u których choroba
pojawiła się w późniejszym wieku, symptomy dzielą się na
łagodne, umiarkowane i ostre. Przebieg choroby jest wolniejszy niż u
dzieci.
Choroba ta może wystąpić zarówno w dzieciństwie jak i w wieku
późniejszym. Pierwsze symptomy w wielu przypadkach to trudność w
chodzeniu, ponieważ jako pierwsze choroba atakuje tkanki i kończyny
dolne. Symptomy te są podobne do wielu innych form dystrofii mięśni.
W związku ze słabnięciem mięśni klatki piersiowej często występują
również problemy z oddychaniem w czasie snu (bezdech), poranny
ból głowy, zawroty głowy w ciągu dnia. Często chorzy odczuwają
brak tchu, są niezdolni do ćwiczeń fizycznych.
Objawy choroby Pompego:
- postępujące słabnięcie mięśni zwłaszcza tułowia i kończyn dolnych
- brak tchu
- zmęczenie
- łatwość oddychania tylko w pozycji stojącej i wyprostowanej
- bezdech nocny
- poranny ból głowy
- zawroty głowy w ciągu dnia
- skolioza
- powiększona wątroba
- powiększony język
- trudność w połykaniu i przeżuwaniu pokarmu
- ból w dole krzyża
Poziom inteligencji pacjenta pozostaje bez zmian; rzadko występują
problemy z sercem; w przypadku skoliozy może być konieczny zabieg
chirurgiczny korygujący, co jednak nie zawsze jest możliwe ze względu
na słabe mięśnie otaczające.
Im wcześniej choroba zaatakuje organizm, tym przebieg choroby jest
bardziej ostry, uciążliwy. Osobom, które zachorują dopiero w
wieku dorosłym choroba mniej doskwiera, wolniej postępuje. Część
chorych pozostaje sprawna przez wiele lat, lecz wielu, by móc
się przemieszczać, potrzebuje wózka inwalidzkiego, kul i/lub
aparatu
oddechowego.
Choroba lizosomalnego gromadzenia LSD dzieli się na ponad czterdzieści
różnych znanych zaburzeń genetycznych spowodowanych błędem w
metabolizmie organizmu Genetyczny defekt powoduje obniżony poziom
enzymu w obrębie lizosomu, na skutek czego cząsteczki stopniowo ulegają
nagromadzeniu.
Choroby grupy LSD występują rzadko (1 na 7,700 urodzeń). Przykłady
tychże chorób: Gauchem, Tay-Sachs, Fabry, Batten, Krabie,
Hurler-Scheie MPS I, Hunter MPS II, Pompego, Niemann-Pick. Większość
tych chorób jest postępująca i zagraża życiu pacjentów.
Leczenie jest w większości przypadków objawowe (z wyjątkiem typu
I Gauchem i Fabry). Choroby te są dziedziczne, występują na skutek
defektu genetycznego (chorzy dziedziczą dwa nieprawidłowo funkcjonujące
geny, po jednym od rodzica). Choroby: gauchem, MPS I, Pompego i
Niemann-Pick są autosomalne.
Komórka jest podstawowym, najmniejszym elementem organizmu. Sama w sobie jest również miniaturowym organizmem pełniącym funkcje zapewniające jej przetrwanie: oddychanie, wchłanianie, wydalanie. Lizosom jest ważnym elementem komórki. Trawi on składniki odżywcze poprzez rozbijanie cząsteczek odpowiednimi enzymami, przyczyniając się w ten sposób do rozwoju komórki, a następnie jej podziału. Poszczególne cząsteczki rozbijane są na prostsze związki, które następnie biorą udział w budowie nowych struktur komórki, zapewniających jej prawidłowe funkcjonowanie w danej tkance i organie. Balans pomiędzy rozbijaniem i tworzeniem nowych cząsteczek musi być zachowany. Materiał, który nie może być strawiony gromadzi się w obrębie lizosomu, zaburzając w ten sposób jego pracę. Spowodowane jest to błędem genetycznym.
Choroba Pompego jest dziedziczna, przenoszona genetycznie z rodziców na dziecko. Geny zawierają instrukcje wszystkich procesów zachodzących w organizmie min. dotyczących produkcji enzymów odpowiedzialnych za rozbijanie glikogenu (GAA). Chorzy dziedziczą zmutowane geny od obojga rodziców, w związku z czym enzym GAA produkowany jest w niewystarczających ilościach lub też nie funkcjonuje prawidłowo. Choroba Pompego jest autosomalna tzn, że gen odpowiedzialny za GAA ulokowany jest na jednej z 21 par chromosomów zwanych autosomami (lecz nie na chromosomach determinujących płeć - dlatego też choroba ta nie zależy od płci) Osoby, które odziedziczyły tylko jedna kopię zmutowanego genu nazywane są nosicielami choroby. Nie obserwuje się u nich symptomów choroby, są zdrowi. Jeżeli jednak dany nosiciel ma potomstwo z drugim nosicielem, wówczas ich dzieci mogą zostać zaatakowane chorobą Pompego. Jeżeli dziecko odziedziczy dwa zdrowe geny, wówczas choroba mu nie zagraża. Są cztery możliwe wyniki 'loterii genowej': dziecko dziedziczy dwa zmutowane geny po jednym od rodzica; dziecko dziedziczy jeden zmutowany gen od matki i jeden zdrowy od ojca; dziecko dziedziczy jeden zmutowany gen od ojca i jeden zdrowy od matki; dziecko dziedziczy dwa zdrowe geny po jednym od rodzica. Jeżeli oboje rodzice są nosicielami choroby, wówczas istnieje:
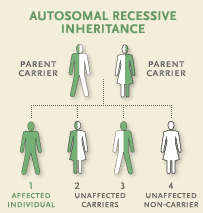
- 25% prawdopodobieństwo (przy każdej ciąży), iż dziecko otrzyma dwa normalne geny i będzie zdrowe
- 50% prawdopodobieństwo (przy każdej ciąży), że dziecko otrzyma jeden normalny gen od matki lub ojca i będzie nosicielem choroby. Dzieci te nie odczuwają żadnych symptomów, lecz mogą przekazać zmutowane geny swemu potomstwu
- 25% prawdopodobieństwo (przy każdej ciąży), że dziecko otrzyma dwa zmutowane geny i zachoruje na chorobę Pompego
- 1932 - Holenderski patolog J.C.Pompę opisuje przypadek siedmiomiesięcznego dziecka, które zmarło nagle z powodu nagromadzenia glikogenu w wielu tkankach.
- 1954 - Laureat nagrody Nobla G.T.Cori odkrył i zbadał przebieg metabolizmu glikogenu i sklasyfikował chorobę jako gromadzenie glikogenu (GSD II).
- 1963 - H G Hers połączył chorobę Pompego z dziedzicznym brakiem lub niedostatkiem enzymów lizosomalnych, klasyfikująca w ten sposób chorobę jako gromadzenia w obrębie lizosomu (LSD).
- 1966 - 70 - A.G.Engel wyróżnia poszczególne typy choroby.
- 1968 - 70 - Pierwsze próby zdiagnozowania choroby Pompego.
- 1979 - Gen odpowiedzialny za występowanie choroby Pompego zostaje przypisany chromosomami 17 i oznaczony jako GAA na ludzkiej mapie genów.
- 1986 - Nieudana transplantacja szpiku kostnego u pacjenta z chorobą Pompego.
- 1991 - Van de Ploeg publikuje pozytywne rezultaty użycia zastrzyków zastrzyków enzymu GAA. 2000 - Van den Hout publikuje pierwsze rezultaty zastosowania terapii enzymowej (ERT).
- 2001 - Amalfitano publikuje ocenę terapii eznymowej (ERT) w Stanach Zjednoczonych.
Objawy choroby występujące u niemowląt to trudności przy karmieniu lub
powiększenie mięśnia sercowego. Starsi pacjenci mogą skarżyć się na
słabość mięśni, zmęczenie lub problemy z oddychaniem. Choroba Pompego
jest łatwiejsza do zdiagnozowania u niemowląt, ponieważ postęp choroby
następuje u nich znacznie szybciej i efekty stają się szybko widoczne.
Diagnozowanie choroby może być przeprowadzone prze wielu różnych
specjalistów jak: pediatra, kardiolog, neurolog, genetyk, itd.
71% przypadków choroby Pompego u niemowląt jest diagnozowanych
przez pediatrów, podczas gdy 80% przypadków u ludzi
dorosłych u neurologów. W przypadku osób, które
zachorowały w wieku późniejszym (nie dzieci) zdiagnozowanie
choroby jest trudniejsze i często przypisywane jest różnym typom
dystrofii mięśni, ze względu na rzadkość występowania choroby Pompego.
Pacjent, u którego istnieje podejrzenie choroby musi mieć
przeprowadzoną biopsje skóry lub mięśni, by zbadać poziom
aktywności enzymu GAA (u większości chorych nie przekracza on 40%
normalnego poziomu). U osób, które są potencjalnymi
nosicielami choroby i planują założenie rodziny, stosuje się
monitorowanie zwane amniocentesis, podczas którego do brzucha
matki wprowadzana jest igła w celu pobrania próbki osłonki płodu
"(pierwszy zabieg miał miejsce w latach 60). Inny sposób
monitorowania (stosowany obecnie) polega na analizie poziomu
enzymów pochodzących z komórek chorionic willi. Metoda to
umożliwia zdiagnozowanie choroby już w dwunastym tygodniu ciąży.
W celu zdiagnozowania choroby wykonuje się: biopsję skóry lub mięśni, badania krwi, elektromiografię, echokardiografię i elektrokardiografię.
- Biopsja skóry lub mięśni: metoda ta polega na badaniu poziomu aktywności enzymów na podstawie analizy pobranej próbki tkanki. Stosuje się znieczulenie miejscowe. Biopsja skóry jest mniej inwazyjna niż biopsja mięśni i bardziej pomocna w diagnozowaniu choroby Pompego. Biopsja mięśni natomiast może potwierdzić zwiększone nagromadzenie glikogenu w mięśniach.
- Badania krwi: badając krew można określić poziom enzymu creatine kinase (CK). 90% chorych ma podwyższony poziom tego enzymu, co wskazuje na zaburzenia pracy mięśni. U pacjentów, którzy mają powiększoną wątrobę bada się również poziom enzymów wątrobowych na podstawie badań krwi (zwłaszcza enzymy: aspartate aminotransferase AST i alaninę aminotransferase ALT)
- Elektromiografia: stosowana z zaburzeniach neuronowych mięśni. Polega na umiejscowieniu elektrod na skórze w celu monitorowania aktywnościposzczególnych mięśni.
- Radiologia (promienie X): prześwietlenie klatki piersiowej służy ocenie mięśnia sercowego i, w przypadku jego powiększenia zaleca się echokardiografię i elektrokardiografię. Służą one do określania czy pacjent cierpi na wczesnopoczątkowy, czy póżnopoczątkowy typ choroby oraz jak bardzo mięsień sercowy jest powiększony, jego grubość i zdolność pompowania krwi.
Choroba Pompego dzieli wiele wspólnych z innymi zaburzeniami:
-
wczesnopoczątkowy typ choroby Pompego.
Chorobna Podobne symptomy Accute Werding-Hoffman Postępujące osłabienie mięśni, brak odruchów warunkowych Danone Powiększenie mięśnia sercowego, nadmierna gromadzenie glikogenu Endocardial fibroelastosis Brak tchu, trudność w karmieniu, powiększenie mięśnia sercowego wada serca Gromadzenie glikogenu III,VI Powiększona wątroba, słabość mięśni, podwyższony poziom creatine kinase (CK) Idiopathic hypertrophic cardiomyopathy Powiększenia obu komór serca Wada wątroby Podwyższony poziom aspartate aminotransferase (AST), powiększenie wątroby Zaburzenia mitochondrialne Powiększona wątroba, choroba serca, podwyższony creatine kinase (CK) Myocarditis Zapalenie mięśnia sercowego prowadzące do jego powiększenia późnopoczątkowy typ choroby Pompego.
Chorobna Podobne symptomy Dystrofia mięśni Kuchenne (DMD) Postępujące słabnięcie mięśni, problemy z chodzeniem Gromadzenie glikogenu III, VI Powiększona wątroba, słabość mięśni, podwyższony poziom creatine kinase (CK) Gromadzenie glikogenu V Podwyższony poziom creatine kinase (CK), skurcze mięśni podczas ćwiczeń Wada wątroby Podwyższony poziom aspartate aminotransferase (AST), powiększenie wątroby Dystrofia mięśnia Limb-girdle (LGMD) Postępujące słabnięcie mięśni w miednicy, nogach i ramionach Polymyositis (zapalenie mięśni) Słabość mięśni niewiadomego pochodzenia Syndrom sztywnego kręgosłupa Sztywność mięśni kręgosłupa, ból w dole krzyża Artretyzm reumatoidalny Bolesność mięśni po wysiłku Scapuloperoneal syndrome Postępujące słabnięcie mięśni w obrębie kolan i ramion Sleep apnea Poranny ból głowy, częste nocne budzenie, zmęczenie w ciągu dnia i zawroty głowy
